User Guide
Triggate
A computational pipeline for designing toehold switches in eukaryotes and prokaryotes
Introduction
The user interface of the tool has a simple mode of operation, where user is required to fill some basic fields and other default values are used. However, for the more advanced user, there is a button 'Show Advanced Options' that lets the user change all other parameters. After filling and submitting the form, our servers using our software tool algorithms will generate a csv file with list of optimized toehold switches candidates according to the specified parameters. Another csv file will be generated containing the sequences of the original expression and trigger genes as well as their optimized version if was required. Finally, a visual report pdf file is generated and all three file are sent to user's provided email address. The switch is designed to optimize translation in environments with the trigger sequence or mRNA expressed.
Conceptually, there are three stages to toehold system design. First Stage is the selection of the trigger RNA molecule, which can be and mRNA or any other non coding RNA sequence. The trigger molecule selected has to exhibit the attribute of being highly expressed in the target cell type or environment where the expression gene of the toehold switch should be expressed, while having low expression in non target cell types or environments. Since this stage requires a diverse databases and different other considerations can be taken, we assume the user already has selected an RNA trigger molecule. Second stage is the selection of appropriate segments in the RNA trigger molecule that are optimized to be trigger for activating the toehold switch. We perform algorithms for finding such segment, however, user may skip that stage in case an already selected trigger RNA segment is provided. The third and final stage is the one which generates optimized toehold switches for specified trigger segment and other constraints.
Web Tool Basic Options
The basic user interface form (illustrated in the following figure) requires the minimal set of the following fields to be filled:
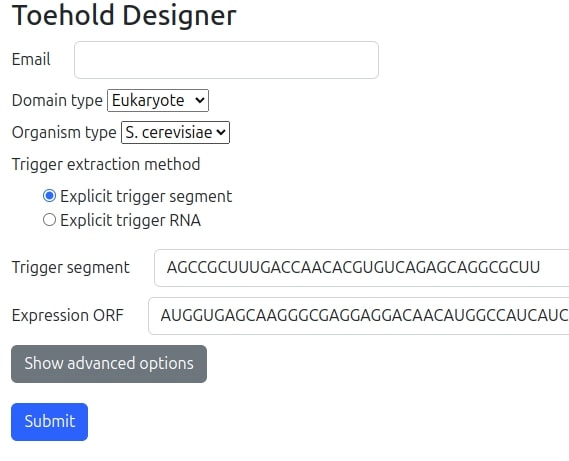
- Email: Email address to send the design report.
- Domain type: The model supports both eukaryotes and prokaryotes. For each different domain, different design architectures will be used and also different dynamic appropriate fields will be required in the advanced form's section according the selected domain.
- Organism type: The model supports a few model organisms for each of the domains. This selection has impact for usage of optimized RBS/Kozak sequence as well as when translation is specified to be optimized appropriate organism database of codon usage statistics will be used and also PSSM table of Kozak first codons. In case 'Customized' option is selected an explicit RBS or Kozak will be required to be filled in the 'Advanced options' section and no translation optimization for CAI and PSSM algorithm will be performed.
- Trigger extraction method: As explained above we can either provide an explicit trigger segment sequence already optimized to be a trigger, thus skipping stage 2 in the pipeline (See flow chart above) by selecting 'Explicit trigger segment' option. However, we may specify a pre-selected mRNA trigger sequence and allow our algorithms to calculate the optimal segment by selecting 'Explicit trigger RNA' option. If 'Explicit trigger RNA' option is selected another sub-form will be dynamically opened as will be illustrated in the next figure.
- Trigger segment: This is explicit trigger RNA sequence to be optimized for.
- Expression ORF: This is the desired expression ORF (Open reading frame of the gene) sequence of the toehold switch to be expressed when in ON state.
- Advanced options: This button dynamically extends the form for many other advanced feature specification and will be elaborated in the following sections.
Trigger Segment Selection Options
When Trigger extraction method is specified to be 'Explicit trigger RNA' the user interface form (illustrated in the following figure) dynamically extends and requires few more fields for the selection of the optimized segment from the whole specified RNA sequence:
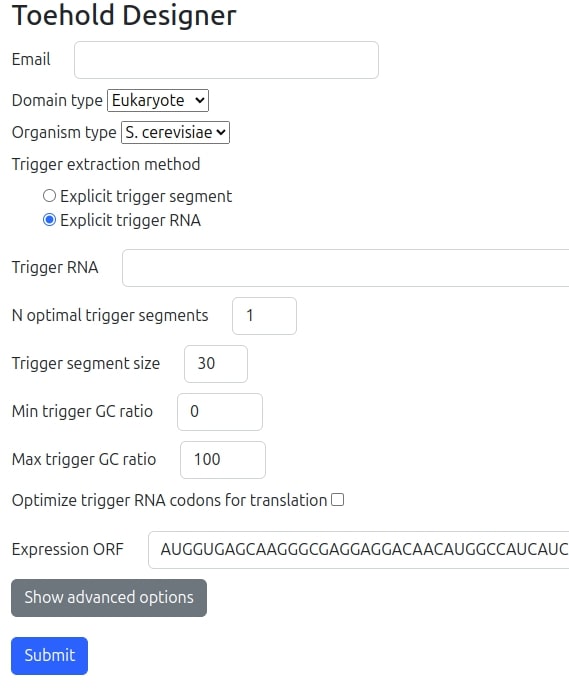
- Trigger RNA: The trigger RNA sequence.
- N optimal trigger segments: The number of optimal trigger segment candidates the algorithm will select and generate a few switches for each of them. The number of seeds at the end of the advanced options sets the number of switches generated for each trigger segment candidate. . The max allowed number of different triggers per single run is 3.
- Trigger segment size: The size of the target segment size to be optimized as a high probable non-folded window/segment in the whole RNA molecule. This field is DISABLED and is only changed IMPLICITLY by the sum of two fields in the advanced options, the trigger binding site size and the opened stem size which when combined should match the reverse complementary segment of the trigger.
- Min trigger GC ratio: We may filter the algorithm to select best segments having minimal GC ratio as specified.
- Max trigger GC ratio: We may filter the algorithm to select best segments having maximal GC ratio as specified.
- Optimize trigger RNA codons for translation: In case the trigger RNA is an mRNA molecule we may optimize its codons for high translation. In that case databases for the organism type selected above with codons statistics will be used.
Advanced Prokaryotes Options
When 'Advanced options' button is pressed and domain type specified is prokaryotes the user interface form (illustrated in the following figure) dynamically extends and allows tuning of many more fields:
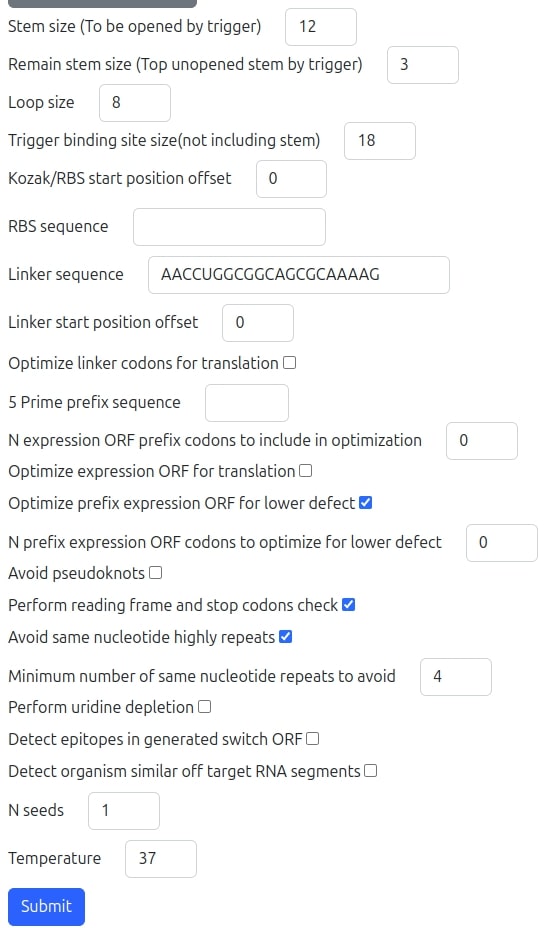
- Stem Size: The size of the hairpin's stem to be opened by the hybridization with the trigger sequence when in ON state.
- Remain stem size: The size of hairpin's stem near the loop's hairpin not designed to be opened by the trigger (In prokaryotes the architecture has a bulge so anyhow there is a remain opened stem but for eukaryotes it can be added.
- Loop size: The size of the hairpin's loop.
- Trigger binding site size: The size of the toehold binding size which is the non-folded switch pre-stem (upstream the stem) segment designed to hybridize with the trigger segment. The trigger also hybridize with segment in the stem with stem size. So trigger size has to be at least the sum of sizes of stem size and trigger binding site size.
- Kozak/RBS start position offset: This is the positive or negative offset of RBS for prokaryotes and Kozak for eukaryotes. If Kozak or RBS are in loop it is by default at the end of the loop so negative offset will move them toward the beginning of the loop but positive offset will move it into the stem and not allowed. In case of Kozak down not in loop positive values are possible to add some non constraint degree of freedom nucleotides after the stem.
- RBS sequence: This is the RBS sequence field which appears only when 'Customized' organism type was selected. If a supported organism type was selected an appropriate RBS sequence will be used.
- Linker sequence: This is the Linker sequence which is needed after the stem when RBS or Kozak are at the loop so translation starts at stem. In that case after the stem a linker is added followed by the ORF of expression gene.
- Linker start position offset: A positive offset may be selected to introduce non constraint degree of freedom nucleotides after the stem before the linker.
- Optimize linker codons for translation: Optimize codons of the linker for high translation. In that case databases for the organism type selected above with codons statistics will be used.
- 5 Prime prefix sequence: A customized conserved sequence to be starting the 5' UTR of the switch. For example 'GGG' are used sometimes for prokaryotes transcription optimization.
- N expression ORF prefix codons to include in optimization: Number of prefix expression ORF codons the algorithm uses in optimizations.
- Optimize expression ORF for translation: Specifies whether tool should optimize expression ORF codons for high translation. In that case databases for the organism type selected above with codons statistics will be used.
- Optimize prefix expression ORF for lower defect: Specifies whether to add degree of freedom and allow the prefix expression ORF codons to be synonymously optimized for achieving lower ensemble defect if possible.
- N prefix expression ORF codons to optimize for lower defect: Specifies the number of allowed prefix expression ORF codons to be synonymously optimized for lower defect. This number should be less or equal to the number specified above 'N expression ORF prefix codons to include in optimization'.
- Avoid pseudoknots: Specifies whether the algorithm directed to try avoiding substantial pseudoknot in the switch design structure.
- Perform reading frame and stop codons check: Specifies whether the algorithm should perform correct reading frame and stop codons validation.
- Avoid same nucleotide highly repeats: Specifies whether the algorithm should not allow switch with same nucleotide repeated over some limit.
- Minimum number of same nucleotide repeats to avoid: Specifies the number of disallowed same nucleotide repeats in the switch.
- Perform uridine depletion: Specifies whether the algorithm should optimize codon for lower number of Uridine nucleotides. This feature is designed for in vivo switches.
- Detect epitopes in generated switch ORF: Specifies whether the algorithm seeks in epitopes Database for a possible RNA orf segment of the generated switch which may activate immune response. This feature is designed for in vivo switches.
- Detect organism similar off target RNA segments: Specifies whether the algorithm seeks in RNA Database for the specified organism an RNA which aligns with each of the trigger segments selected and is therefore a possible off target. The effect is also a result of the similar RNA abundance which may be checked further by the user if such warning exists in our report.
- Number of seeds: Specifies the number of different random seeds repeated runs the algorithm should perform. Due to the randomness of the design core algorithm, different random seeds lead to different optimized switches. We then return all the candidates, each with the ensemble defect but the visual pdf report will be generated only for the lowest defect candidate switch. The max allowed number of different seeds per single run is 7.
- Temperature: The temperature of the experiment's environment.
Advanced Eukaryotes Options
When 'Advanced options' button is pressed and domain type specified is eukaryotes the user interface form (illustrated in the following figure) dynamically extends and allows tuning of many more fields. Most of them where already explained in the section for advanced prokaryotes options and here we will only elaborate on the special field which are unique for eukaryotes selection:
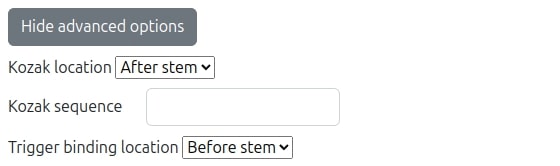
- Kozak location: This specifies which architecture design switch to use, wither one with Kozak 'In loop' which mimics RBS in loop for prokaryotes or use kozak location 'After stem' which is our de-novo design placing the Kozak after the stem and thus also avoiding the usage of linker. In the case of 'After stem' option since linker is not used, all the linker related options will not appear in the form.
- Kozak sequence: This is the Kozak sequence field which appears only when 'Customized' organism type was selected. . If a supported organism type was selected an appropriate Kozak sequence will be used.
- Trigger binding location: This specifies another new architecture for binding the trigger either 'Before stem' options to the pre-stem segment as usual or 'After stem' option for trigger binding to the post-stem segment.
Results Report
Following the user's submission of a job it will be sent to our servers. Our servers will run the job and algorithms will generate candidate switches. Time may vary from few minutes to an hour or even more depending on the complexity and load. In general a longer switch including longer parts(e.g., trigger, stem, and loop) will take longer to optimize. Applying special optimization will take longer, for example applying the codon optimization for lower defect will double and triple the optimization time depending also on the number of codons specified to be taken into the optimization. The waiting time of may grow proportionally to the multiplication of the number of different triggers with the number of seeds candidates which will be generated for each of the triggers. So when servers are in high load job will not enjoy parallelism and upper limit for expected time is (T_single_switch * seeds * n_triggers).
When the job is done and all switch candidates are generated result files will be sent to the user via the provided email address. Two CSV files with metadata will be attached and a few visual pdf report files for each of the different triggers with one best seed. Each of the file names will be prefixed by a UUID unique code.
- Genes metadata file: a CSV file named 'web_genes_info', containing the original expression gene and trigger gene if used and their and optimized version in case codon optimization was required for translation and/or uridine depletion.
- Toehold switch results file: a CSV file named 'web_raw_results', containing a list of all the toehold switch candidates, each generated by using a different random seed, and their generation parameters specified and some metadata.
- Visual report and summary file: a PDF file with an encoded name, containing a summary of up to 2 best candidates having the lowest defect score and predicted structural visual image and two dot plot one for toehold switch in OFF state without the trigger segment and another one in ON state with the trigger.
We will list and elaborate on the most relevant fields of each file
Genes metadata file
- org_trigger_gene: Field with the original trigger gene sequence in case explicit trigger RNA was provided and not already pre-selected segment.
- opt_trigger_gene: Field with the optimized trigger gene sequence in case explicit trigger was used and translation optimization or uridine depletion were required.
- org_reporter_gene_without_aug: Field with the original expression (reporter) gene without the AUG start codon since AUG is included in Kozak for eukaryotes and set after RBS for prokaryotes.
- opt_reporter_gene_without_aug: Field with the optimized expression gene in case codon optimization for translation or uridine depletion was required.
- org_linker: Field with original linker sequence.
- opt_linker: Field with optimized linker sequence in case codon optimization for translation or uridine depletion were required.
Remark: Translation optimization for each of the trigger gene, expression gene and linker has its own optimization flag, but the uridine depletion has single flag and performs depletion for all the three if selected.
Toehold switch results file
The CSV results file will contain the number of seeds specified for each different trigger from n_triggers requested, thus in total (n_triggers * seed) rows, each bunch of rows with the same trigger has a different candidate switch result generated by a different random seed. We explain general meaning of the metadata for each column name of the csv file:
- 'switch': Field with the optimized toehold switch sequence. This is the actual output all other fields are the parameters filled in form used to generate the switch and some debug and metadata information.
- 'temperature': Field with the filled form 'temperature' parameter.
- 'batch_desc': Field with inner metadata for web is const 'web_run'.
- 'trigger_bind_strategy': Field with the filled form 'Trigger binding location' parameter.
- 'trigger_binding_site_size': Field with the filled form 'Trigger binding site size' parameter.
- 'trigger_gene_offset': Field with the offset of the selected optimized segment in the trigger gene if explicit trigger RNA specified. In case explicit trigger segment was provided it will be set to -1.
- 'full_trigger': Field with trigger segment provided or the trigger segment calculated and having size Trigger segment size filled.
- 'full_trigger_gc': Field with a value of the GC ratio of the full trigger.
- 'trigger': Field with sequence equal or prefix of the full trigger, since the actual size used will be the sum of Trigger binding site size and stem size, if the sum is longer it is an error but if shorter only prefix is used.
- 'trigger_gc': Field with a value of the GC ratio of the trigger.
- 'trigger_off_target': Field with information regarding existence of an off target RNA sequence similar to the selected trigger compared by blast with RNA database for the specified organism. If option for checking off targets was not specified it will return 'Not Checked', otherwise it will either return 'None' when not found or a description of the highest hit RNA.
- 'kozak_location_strategy': Field with the filled form 'Kozak location' for eukaryotes parameter.
- 'remain_stem_size': Field with the filled form 'Remain stem size' parameter.
- 'stem_target_energy': Field with metadata regarding stem binding energy.
- 'stem_estimated_energy': Field with metadata regarding estimated stem binding energy.
- 'stem_size': Field with the filled form 'Stem size' parameter.
- 'loop_size': Field with the filled form 'Loop size' parameter.
- 'kozak_start_pos_offset': Field with the filled form 'Kozak/RBS start position offset' parameter.
- 'linker_start_pos_offset': Field with the filled form 'Linker start position offset' parameter.
- 'gene_opt_size': Field with the size in nucleotides that were used from prefix expression gene for structural optimization which is the same as the filled 'N expression ORF prefix codons to include in optimization' parameter multiplied by 3.
- 'gene_opt_start_offset': Field with the offset in nucleotides to start lower defect optimization in the expression gene without AUG, in most cases where translation optimization was required and PSSM for first codon after AUG was performed it will be set to 3, meaning the lower defect optimization was instructed skip first codon after AUG and not to change its already translation optimized first codon.
- 'gene_opt_end_offset': Field with the offset in nucleotides to end lower defect optimization in the expression gene without AUG, this is the same as the filled 'N prefix expression ORF codons to optimize for lower defect' parameter multiplied by 3.
- 'use_codon_bias': Field with the filled form 'Optimize prefix expression ORF for lower defect' parameter.
- 'is_run_pseudoknots': Field with the filled form 'Avoid pseudoknots' parameter.
- 'is_avoid_repeats': Field with the filled form 'Avoid same nucleotide highly repeats' parameter.
- 'n_avoid_repeats': Field with the filled form 'Minimum number of same nucleotide repeats to avoid' parameter.
- 'less_important_factor': Not used in web interface operation mode.
- 'kozak': Field with the filled form 'Kozak sequence' parameter.
- 'prefix': Field with the filled form '5 Prime prefix sequence' parameter.
- 'linker': Field with the filled form 'Linker sequence' parameter.
- 'unopened_stem_seq': Not used in web interface operation mode.
- 'seed': The actual random seed value used for debug purposes.
- 'reporter_gene_without_aug': The same sequence also in the genes metadata file.
- '####_strand': Metadata fields for debug.
- '####_structure_optimal': Metadata fields for debug.
- 'switch_domain_codes': Metadata fields used for colors of different RNA subcomponents.
- 'ensemble_defect': The main score we use for switch generation and the 2 candidates with that lowest score are chosen for the summary and visual report.
- 'tubes_defect': Metadata with elaborated ensemble defects of each used tube and complex in our modeling of the toehold reaction divided to structural defect and concentration defect.
- 'concentrations': Metadata with elaborated concentrations of each used tube and complex in our modeling comparing actual vs target concentrations.
- 'homo_dimer_energy': Metadata with calculated binding energies of a pair of switches composing a homo dimer molecule calculated by two different algorithms by concatenation-based approach algorithm and by accessibility-based approach algorithm.
- 'detected_epitope': Metadata with a sequence of epitope found in the ORF of the switch in case it was specified to be checked in the form, otherwise returns 'Not Checked'.
Visual report and summary file
The PDF contains results for two best candidates of generated toehold switches having the lowest ensemble defect score. Each of the two reports contains two pages of summarized results and metadata with the same data as in the results csv file. Then it contains 2 dot plots of the probability matrix of the switch by itself in predicted OFF state and switch with the trigger in predicted ON state.
We list here two added result fields which appear in the second pdf page of each candidate and do not appear in the CSV metadata fields:
- pk_open_indices_no: This is the number of pseudoknot in the predicted MFE structure (most probable structure according to the used prediction model). This field can be checked in two algorithm mode of operations one when instructed to apply the pseudoknot avoidance and one without.
- n_gene_codon_changed: This is the number of codons actually changed from their original ones in order to achieve lower defect score.
We present here an example of the images added in the visual report:
The first image is a dot plot of the co-folding nucleotides probability matrix of the switch by itself.
Switch co-fold dot plot
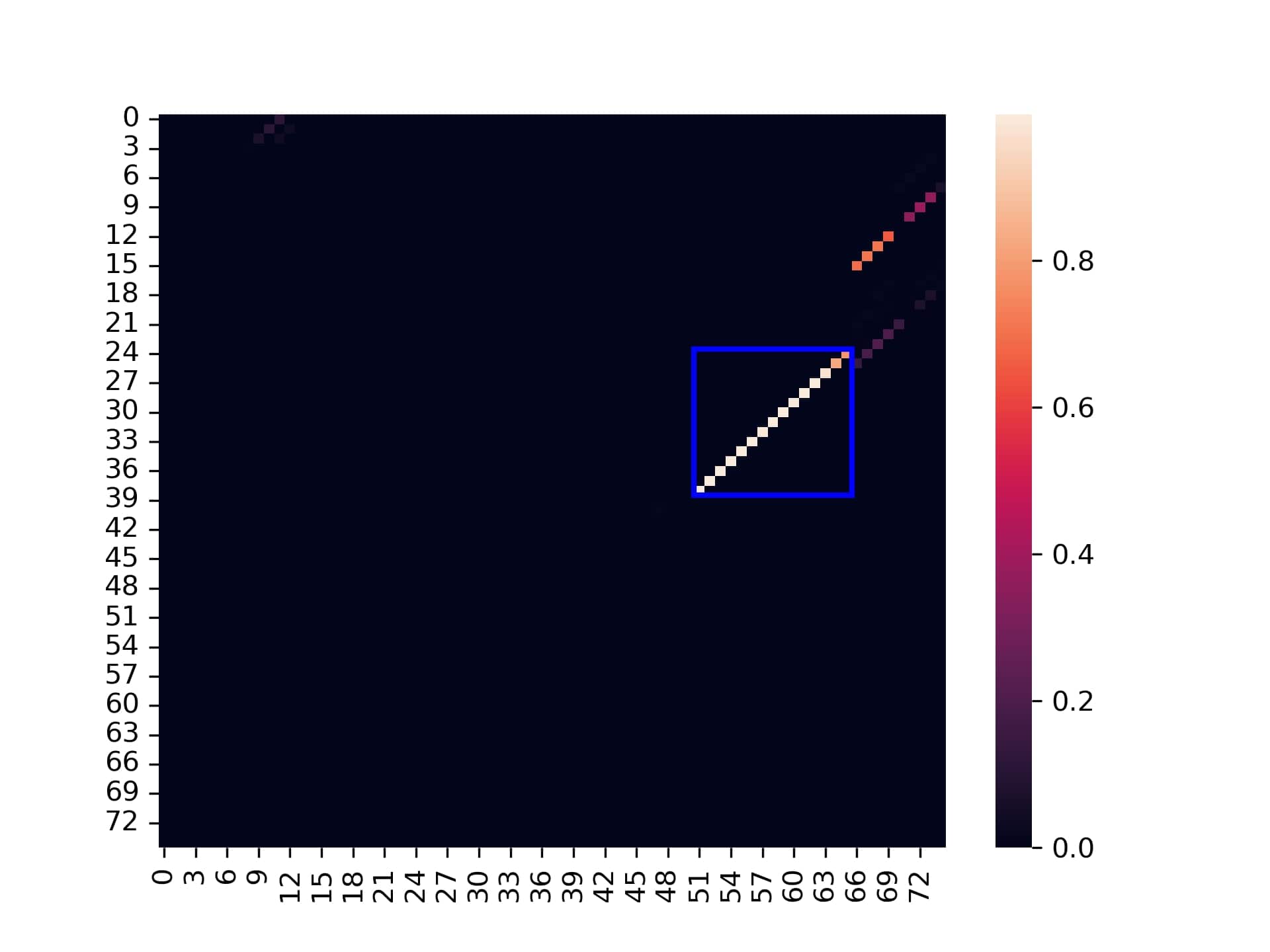
- The blue rectangle: This is the designed stem area which should be bound when the switch is by itself in OFF state. We can see it is very high probable to be self bound area, with few minor other areas "leakage" of probability.
The second image is a dot plot of the co-folding nucleotides probability matrix of the switch and the trigger.
Switch and trigger co-fold dot plot
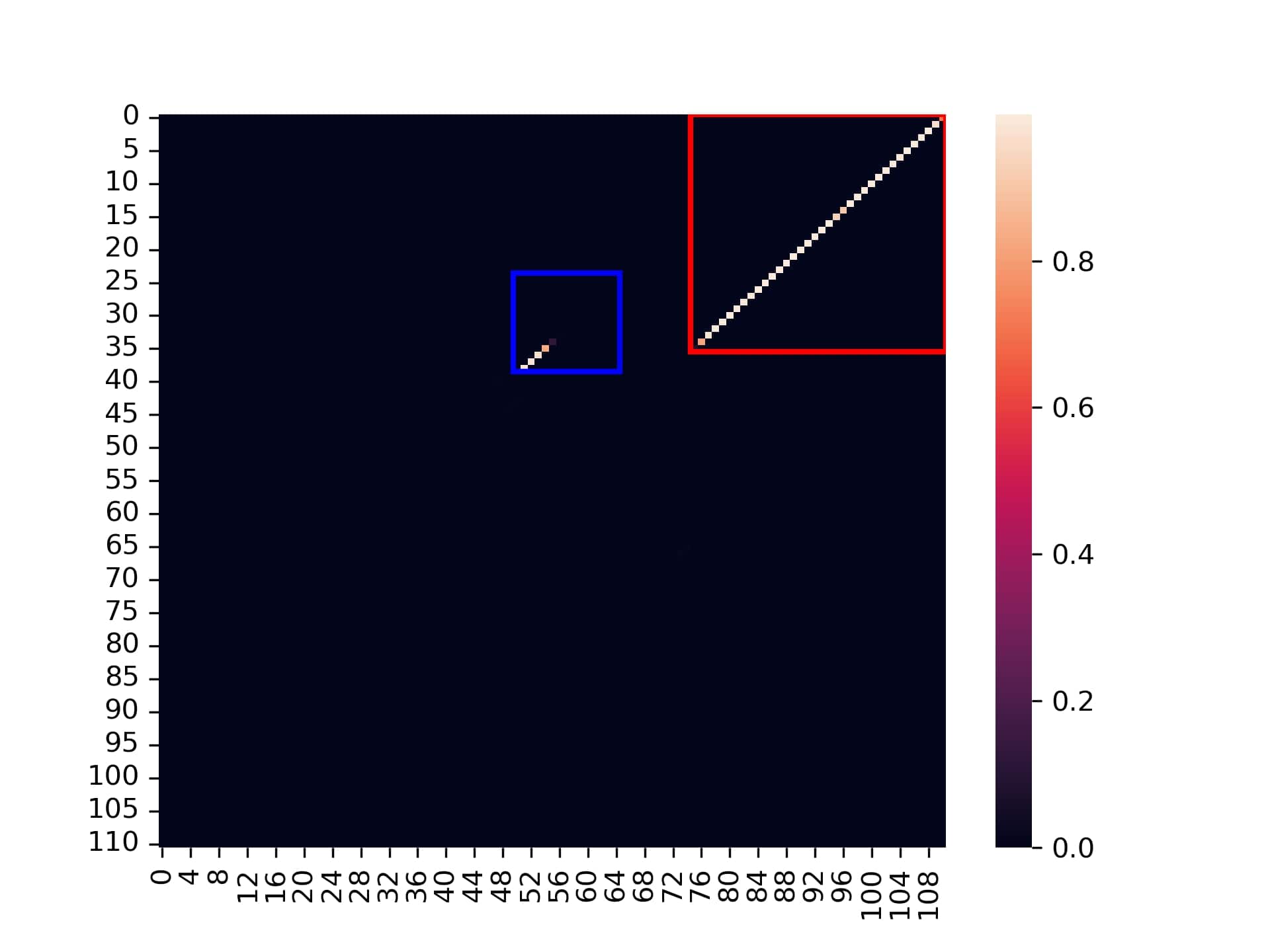
- The blue rectangle: This is the designed stem area which should be unbound when the switch is with the trigger in ON state. We can see it is very high probable to be unbound area, except short line at the diagonal edge which is the desired remain opened stem segment.
- The red rectangle: This is the designed toehold segment in the 5 prime edge of the switch with area that should be bound when the switch is with the trigger in ON state. We can see it is very high probable to be bound area, with almost none visible "leakage" of probability.
The Third image is an RNA the folding MFE secondary structure prediction including pseudoknots of the switch itself in OFF state.
Switch MFE structure with pseudoknots
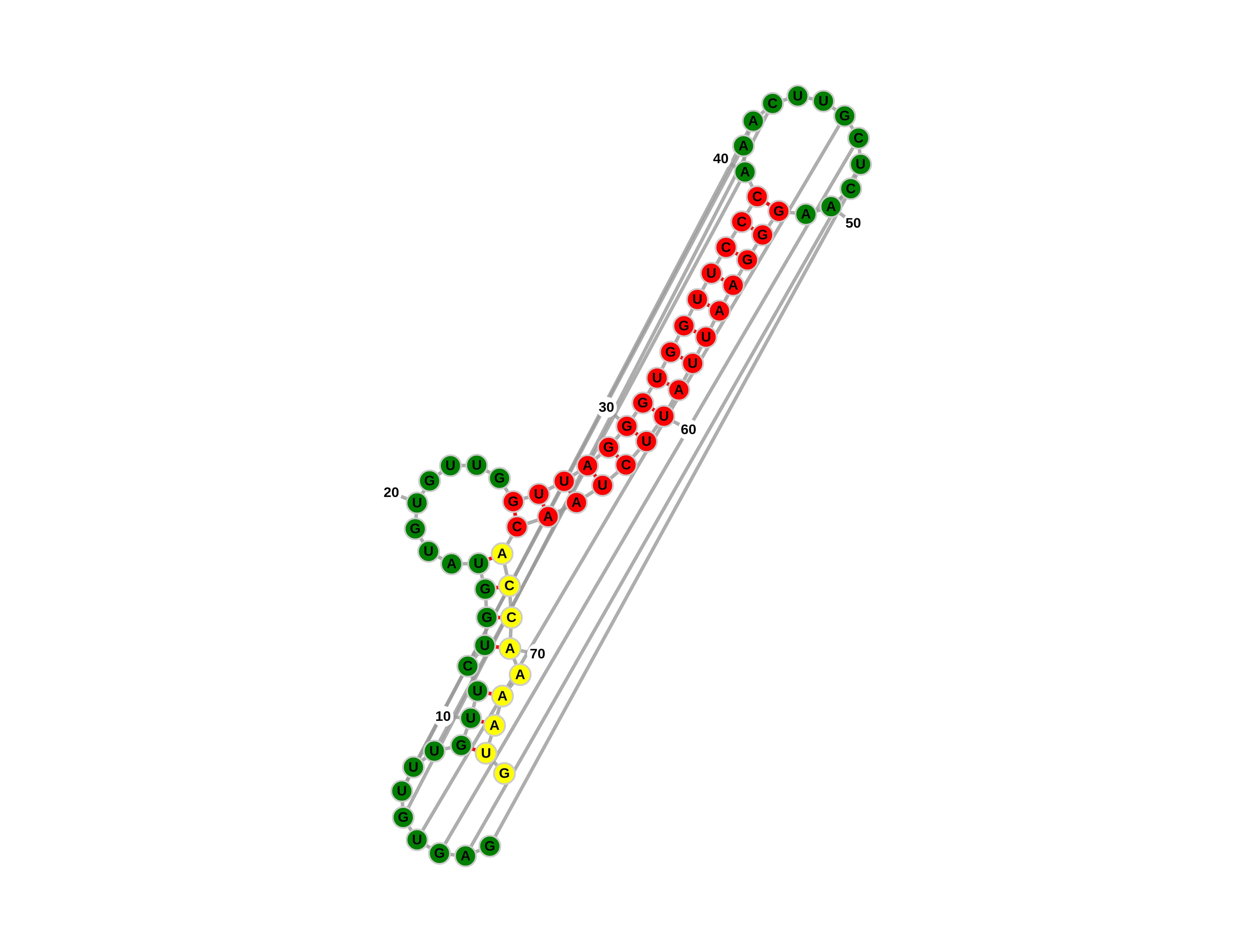
- The different colors: Green marks expected self non-folded segments, red marks expected self folded segments, and yellow is the conserved Kozak sequence.